 [/frame][/lightbox]
[/frame][/lightbox]
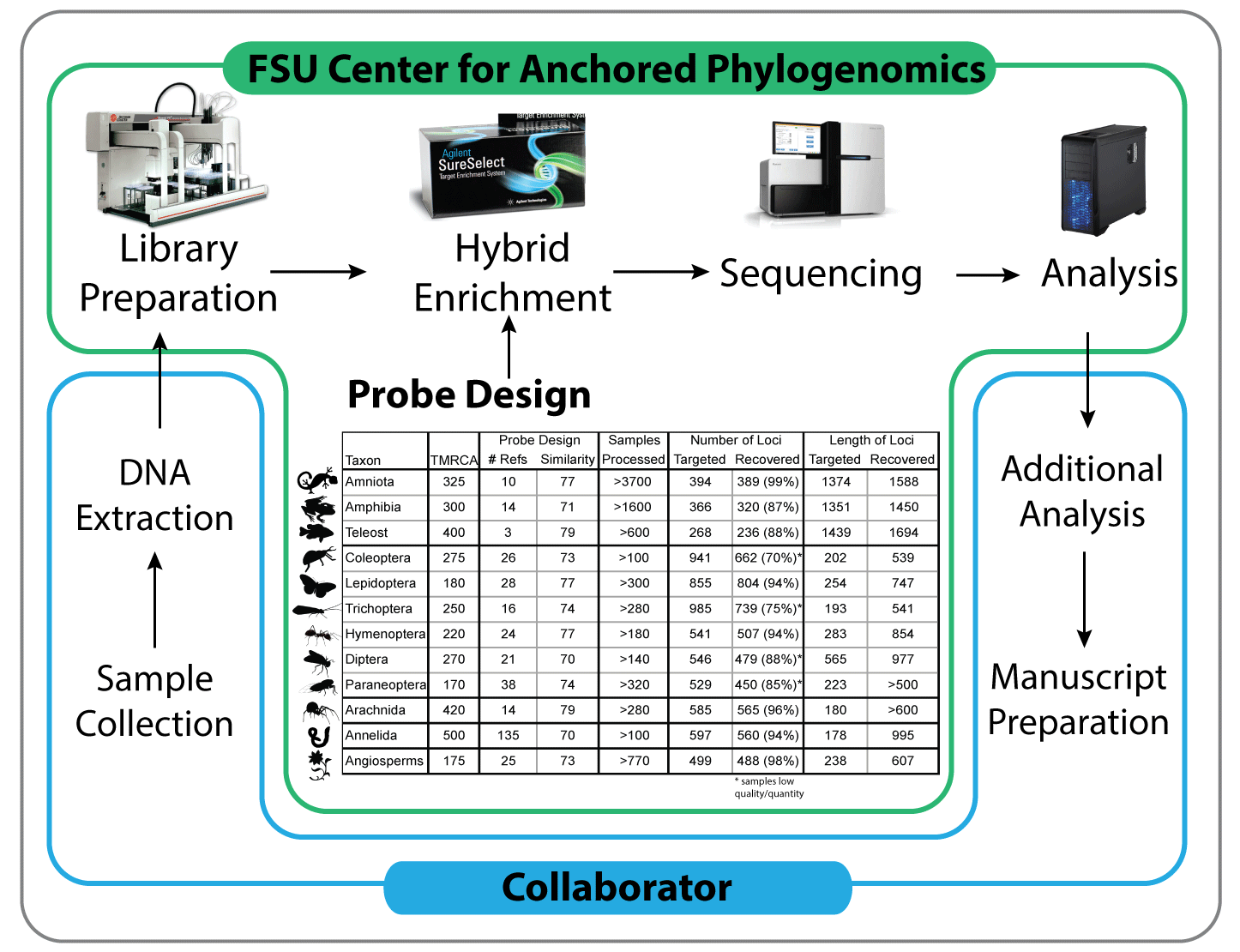
A partnership between researchers and the Center for Anchored Phylogenomics minimizes the cost and processing time associated with data collection, while maximizing the quality and impact of the resulting data. Collaborators extract DNA, perform initial QC and send the samples to the Center. After performing quality control, the Center prepares the samples using a high-throughput liquid-handling robot, enriches the samples for anchor, anonymous, and/or functional loci, sequences the enriched libraries, and performs the bulk of the downstream bioinformatics (including assembly, orthology, alignment, and preliminary phylogeny estimation). The collaborator performs additional analyses and prepares manuscripts for publication in consultation with the Center.
By collaborating with the Center, collaborators benefit from greatly reduced costs that result from the substantial reagent and sequencing discounts that the Center is able to negotiate because of the high volume of samples processed. The Center for Anchored Phylogenomics is a non-profit entity that operates at cost.
[spoiler title=”1. Project Planning”] Initial Contact—We welcome new collaborations. If you would like to discuss a potential project with us, please submit a new collaborator form. Once we receive the request we typically arrange a Skype or phone conference to discuss the potential project. Note that for new taxonomic systems in which we have not yet published, we operate on a first-come first-served basis. A list of current taxonomic systems is listed here. The first collaborator(s) working with us on a new system typically invests in extra genomic resources necessary to ensure success of the anchored phylogenomics approach in that system. Thus, we are careful to coordinate with the initial collaborator before starting new collaborations with other researchers on that system to avoid overlap. Once the first publication in a new system is accepted and online, the system is open for additional collaboration without approval of the initial collaborator. We make every attempt to avoid conflict and encourage cooperation among collaborators working on similar systems by facilitating joint projects when possible. For all collaborations, we conduct the data collection and analysis at cost in exchange for co-authorship (Alan Lemmon and Emily Moriarty Lemmon) on relevant papers.Define Scope—During initial conferences with collaborators, we discuss the taxonomic breadth, divergence times of deepest lineages in the clade, and projected number of samples to be processed. We determine whether one of our existing probe sets can be used for the project or if a custom probe set should be designed. In the latter case, we identify whether it is necessary first to obtain additional genomic resources for the system for designing more specific probes (to improve efficiency of data collection). We also discuss collaborator timelines (e.g., upcoming grant deadlines for which preliminary data are needed), expected manuscripts, target audience, etc.
Identify Challenges and Solutions—Although the majority of projects are straightforward, some biological systems present new challenges, such as very large genome sizes (>5Gb), small starting DNA quantities (<200ng), low DNA quality (e.g., degraded samples), insufficient genomic resources, and abundance of introns. We have developed successful strategies for overcoming many of these challenges and are continually testing new ways to improve efficiency of data collection and analysis.
Estimate Project Costs—After outlining the project with the collaborator and developing a strategy for moving forward, we provide a specific quote for the cost of the project to the collaborator. After the quote has been approved by the collaborator, we set up a contract, which is submitted to FSU’s Office of Sponsored Research. This office communicates with the collaborator’s institution to facilitate transfer of funds. Please note that all projects are done at cost, since our laboratory at a state university is not a commercial enterprise.
[/spoiler] [spoiler title=”2. Sample Preparation”]DNA Extraction—Genomic DNA is typically extracted by the collaborator and shipped to our laboratory. In some cases we can arrange to do the extractions in our lab (but collaborator will cover costs of extractions — inquire for more information). Samples should be extracted using a Qiagen DNEasy kit (or equivalent) and eluted into Tris EDTA, Tris, Qiagen EB, or equivalent (pH should be between 8 and 9). Please ensure DNA has been treated with RNAse to remove RNA (part of Qiagen kit protocol). RNAse treatment is required before we will process samples. See supporting information for quality of samples with various RNAse treatments.
Initial DNA Quantity—The collaborator should quantify all samples using a Qubit fluorometer whenever possible. If a Qubit is not available, the Nanodrop Spectrophotometer system can be used to ensure 2.0 ug of DNA per sample in 130 microliters of buffer. See supporting documents explaining inconsistencies between these two instruments. In systems where smaller DNA quantities are obtained, collaborators should send as much DNA as possible, without pooling individuals. To date, we have successfully obtained data from samples down to ~50 ng starting DNA. For samples with sub-optimal DNA quantities, we can often obtain sufficient DNA by performing whole-genome amplification prior to starting the library preparation protocol. Note that samples containing <200ng of total DNA may be lost entirely if library preparation is not successful on the first attempt. Please consult with us about any samples with quantities below 2.0 ug.
Initial DNA Quality—The preferred quality of each sample is >1.8 as measured by both 260/280 and 260/230 ratios on a Nanodrop. Samples with lower ratios may require ethanol precipitation (See supporting documents for suggested EtOH protocol) or other treatment to remove salts and other contaminants and improve readings (see Nanodrop manual for explanation). High molecular-weight DNA is strongly preferred, however, degraded samples (fragmented DNA) can still be processed with slight adjustment to our protocol.
Prior to shipping samples we require the collaborator to run an agarose check gel of all DNA samples and email us an image of the gel so that we can assess levels of DNA degradation. Samples should be prepared by combining 4µLsample with 2µL of 6X loading dye. The sample should then be loaded on a 2% agarose gel in 1x TAE and run for 90min at 120V with a ladder that includes fragments from at least 100bp–1000bp. Gels should be labeled using Adobe Illustrator or a similar program to indicate sample position. An example of suitable and unsuitable results is given here. The collaborator will need to find replacements for samples of “Bad” quality as they will not be suitable for library preparation. If alternatives are not available, please consult with us before shipping these poor quality samples. A labeled gel photo in .pdf format should be e-mailed to us for inspection by our technicians prior to shipping samples.
[/spoiler] [spoiler title=”3. Locus Selection and Probe Design for Anchored Hybrid Enrichment”]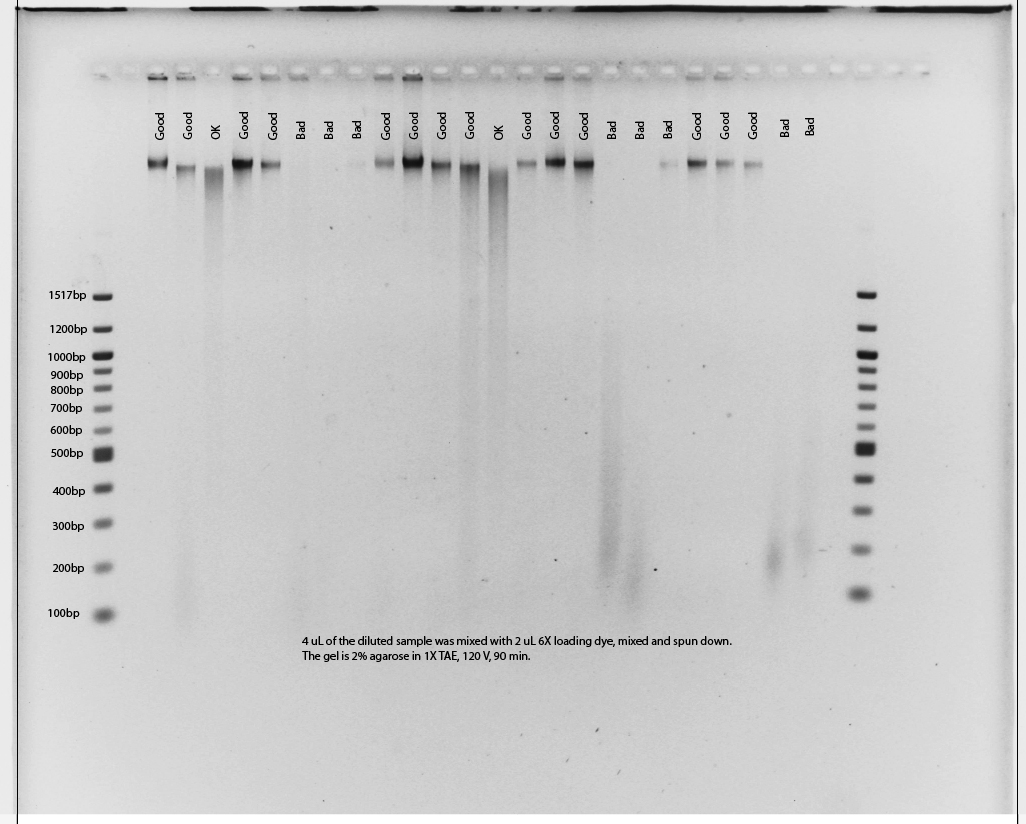{kind=link}
Identify Available Genomic Resources—One key to the success of the anchored phylogenomics approach is the use of multiple reference species during locus selection and probe design, which allows a researcher to target loci with sufficient phylogenetic information for nearly all taxonomic scales. Before initiating a project, we ask collaborators to summarize the current phylogeny of the group with an indication of the lineages for which genomic resources are available (i.e., assembled genomes, transcriptomes, or raw genomic reads), as well as genome sizes (see http:/genomesize.com or http://data.kew.org/cvalues/). This information should be emailed to us as a figure (a rough illustration or even hand-drawing is fine). If divergence times are known (even approximately), please include this information on the phylogeny figure.
Select Target Loci—Candidate target loci can be identified using genomic resources (genomes, transcriptomes, raw genomic reads, etc.) from two or more species. We have experience designing locus sets containing loci of several different types. Core loci are loci that are shared across large taxonomic groups, typically at the phylum level. For example, we have identified ~400 long, single-copy, low indel, nuclear loci (i.e., exons) with sufficiently high levels of sequence variation for most phylogenetic questions across Vertebrata. Identification of these core loci is important because it facilitates rapid data collection in large numbers of species (since core loci only need to be identified once per phylum) and allow for a high degree of integration of results from different studies. In a similar way, some find it beneficial to also target loci that have already been sequenced in large numbers of organisms (e.g., barcoding genes).
Collect Additional Data—Once candidate loci are identified, we determine (in consultation with the collaborator), whether genomic resources already exist for a sufficient diversity of reference species, or whether additional data need to be collected. If necessary, we identify key unrepresented lineages and collect Illumina HiSeq data at 15X coverage for each lineage (note that our bioinformatics pipeline works from raw genomic reads and thus does not require genome assembly). Collaborators provide the genomic DNA, we cover the library preparation costs, and collaborators cover the cost of sequencing at FSU at in-house rates. We perform Paired-End 150bp rapid run lanes on the HiSeq2500, which generates ~40-50Gb per lane. We typically obtain the raw sequencing reads ~2 months after receiving the DNA from the collaborator. For large genomes (>10Gb), the collection of RNA-Seq data may be preferable, although we currently do not offer RNA-Seq library preparation.
Isolate Reference Sequences—After sufficient genomic resources are obtained, we isolate sequences for the target loci for each reference species to be included in the kit design and generate an alignment for each locus. A substantial amount of quality control is applied to these alignments to ensure that the target loci are single copy and have the appropriate amount of sequence variation to ensure phylogenetic accuracy and efficient enrichment.
Order Probes—Once the probes have been designed, we order an enrichment kit from Agilent Technologies, Inc. and have it shipped to our lab. Kits come in two sizes, 96-reactions (~1200 samples) and 16-reactions (~200 samples). We have been able to obtain a large-volume discount on both sizes of kits from Agilent that reduces the price of enrichment substantially. For collaborators working in new systems, we may ask them to purchase the initial 16-reaction kit (at the discounted rate) that will be used for their project. Once the pilot project is complete (paper is accepted for publication), collaborators who invested in the initial kit choose between one of two options: 1) keep the kit in our lab for future projects or 2) have the remainder of the kit sent to their lab. Collaborators are also given details about the probe design at this time. Collaborators working in systems in which we regularly work (e.g., vertebrates) can use reactions from our stock of enrichment reagents at cost. **Note that after we finish a new probe design, enrichment kits require 4-6 weeks for production by Agilent. Re-orders of existing kits are typically faster.
[/spoiler] [spoiler title=”4. Enrichment”]This step is determined on a project-to-project basis after initial discussion and project design. Additional information will be provided to collaborators as a part of this discussion. Please contact us for further inquiry.
[/spoiler] [spoiler title=”5. Sequencing”]Sequence Enriched Libraries—We deliver the enriched libraries to the sequencing center at FSU for final quality control, which includes library concentration estimation using Kappa qPCR and library size analysis on the Agilent Bioanalyzer. After passing final QC, the project enters the sequencing queue. Sequencing is performed on the HiSeq2500. We typically sequence using PE150bp RapidRun mode in order to shorten wait time. A separate 8-bp indexing read is performed. Collaborators may monitor the progress of their runs using Illumina’s BaseSpace Cloud.
[/spoiler] [spoiler title=”6. Analysis of Raw High-Throughput Sequencing Data”]This step is determined on a project-to-project basis after initial discussion and project design. Additional information will be provided to collaborators as a part of this discussion. Please contact us for further inquiry.
[/spoiler] [spoiler title=”7. Publication”]Manuscript Preparation—Collaborators are responsible for writing/publishing one or more manuscripts based on the biological results of the study. The Lemmons will provide the text for the methods relating to the anchored phylogenomics data collection and analysis. The collaborator will be responsible for uploading data files to the database referenced by the publication. The Lemmons may publish one or more additional paper(s) describing the application of the anchored phylogenomics approach to the system under study (without details related to the biological system).
Authorship—Alan Lemmon and Emily Lemmon request authorship on the papers resulting from collaborations. Additional authors may be added after agreement by all parties involved in the collaboration. Authorship order will be decided by the collaborator for the biology-oriented paper(s) and by the Lemmons for the method-oriented paper(s).
[/spoiler] [spoiler title=”8. Literature Cited”]- Ané C, Larget B, Baum DA, Smith SD, Rokas A. 2007. Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 24:412–26
- Lemmon AR, Emme SA, Lemmon EM. 2012. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 61:727–44
- Lemmon EM, Lemmon AR. 2013. High-throughput genomic data in systematics and phylogenetics. Ann. Rev. Ecol. Evol. Syst. 44:19.1–19.23
- Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols 2010(6); doi:10.1101/pdb.prot5448
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–74
- Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21:456–63